Research
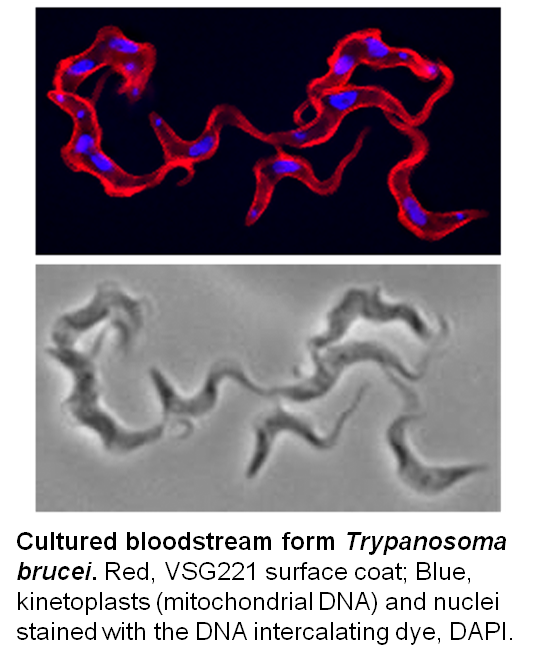
T. b. gambiense and T. b. rhodesiense cause Human African Trypanosomiasis (HAT or sleeping sickness) in West/Central and East Africa, respectively. Though the duration and progression of the two forms of the disease varies, both forms of HAT are typically fatal without treatment (see WHO website and the blog post, ‘Why study African trypanosomes‘). The related but non-human infective T. b. brucei, T. congolense and T. vivax cause the livestock wasting disease, N’agana, which is endemic to sub-Saharan Africa where it has a significant impact on food production. All the African trypanosomes are transmitted by the bite of the tsetse (Glossina spp).
There are several virulence factors that promote trypanosome infection in mammals, including: antigenic variation (enables the parasite to persist in the bloodstream indefinitely, or until the host succumbs); resistance to human serum trypanolytic factors by T. b. gambiense and T. b. rhodesiense (these lyse the non-human infective trypanosomes); differentiation, which allows the trypanosome to limit its population growth, as well as to adapt its physiology in preparation for transmission to the tsetse fly vector; and, emergent drug resistance, which is affecting the efficacy of the limited set of difficult to administer toxic drugs currently available.
In contrast to the African trypanosomes, Leishmania spp. are endemic throughout the Tropics, where they are spread by sand flies and cause a spectrum of disease, from self-limiting cutaneous forms to the highly disfiguring mucocutaneous and the often fatal visceral leishmaniasis (see WHO website). In common with the trypanosomes, treatment is challenging due to drug toxicity and emerging resistance, and our understanding of drug mode-of-action and drug resistance mechanisms is limited. Until recently, these parasites were regarded as less genetically tractable than T. brucei due to the absence of the RNAi machinery and other challenges. However, the application of CRISPR/Cas9 technology has revolutionised the field.
In addition to being parasites of medical and veterinary importance, T. brucei and Leishmania are extremely divergent eukaryotes. Classified as members of the Kinetoplastida, a group within the eukaryotic supergroup, the Excavata, trypanosomes possess many features that are strikingly different to the more widely studied Opisthokonta, such as yeast, C. elegans, Drosophila, Xenopus and mammals. For example, T. brucei is unique amonsgt eukaryotes in its use of RNA polymersase-I to transcribe a subset of its mRNAs. Understanding their biology in relation to these model organisms can inform our understanding of the last eukaryotic ancestor, as well as the constraints and flexibility evidenced by the different evolutionary paths taken in the development of particular features.
Work in the lab is focused on two areas…
Understanding the parasite’s interaction with drugs (Leishmania and T. brucei) and innate immune factors (T. brucei), in particular their uptake and intracellular transit. This has focused our attention on the endo-lysosomal system, its contribution to toxin uptake and transit, and its functional regulation.
Understanding the regulatory cross-talk between the two competing demands for RNA polymerase-I in bloodstream-form trypanosomes: rRNA and VSG mRNA. Significant progress has been made in understanding the regulation and maintenance of monoallelic VSG expression, which underpins antigenic variation in T. brucei, but comparatively little is known about rRNA transcriptional regulation.
Specifically…
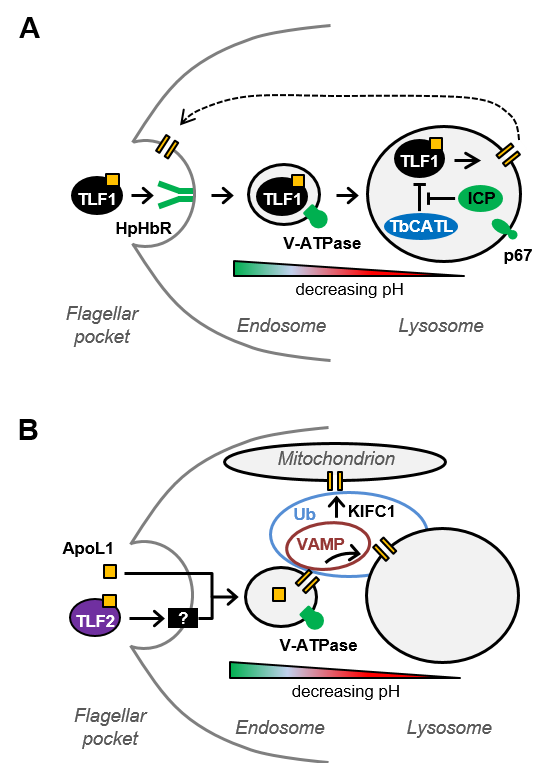
1. Having identified 63 proteins that contribute to the anti-trypanosomal action of apoL1, including multiple ubiquitin modifiers and several membrane trafficking proteins, we are now exploring the contributions of these proteins to apoL1 sensitivity and to endo-lysosomal function. (Currier et al, PLoS Pathogens, 2018)
2. Identifying anti-kinetoplastid drug efficacy determinants, i.e. what parasite proteins drive drug action, including transporters, activators and their associated regulatory networks. This chemico-genomics approach can also lead to insights into protein function beyond roles in drug action. (Collett et al, Antimicrobial Agents and Chemotherapy, 2019)
3. Understanding the regulation of rRNA transcription and how this intersects with RNA polymerase-I transcription of the major surface antigen mRNA in bloodstream-form (VSG) and insect-stage (procyclins) T. brucei. Only a subset of rDNA arrays are transcriptionally active in BSF T. brucei – we are developing the molecular tools to enable us to decode the protein networks responsible for this transcriptional repression and to explore their impact on RNA polymerase-I mediated VSG transcription.
Applying forward genetics in T. brucei to understand trypanosomatid biology
We use a genome-scale RNAi library to identify previously unkown factors that contribute to these areas of trypanosome biology. This methodology has previously been used to identify the genes/transcripts required for growth in culture as bloodstream, procyclic or differentiating cells (Alsford et al, Genome Research, 2011).
The RNAi library can also be used to identify parasite protein networks that contribute to a trypanocidal agent’s efficacy. We initially applied this approach to the five anti-HAT drugs currently in use (Alsford et al, Nature, 2012) and more recently used human serum and apoL1 selection to identify 63 apoL1 sensitivity determinants (Alsford et al, PLoS Pathogens, 2014; Currier et al, PLoS Pathogens, 2018), leading to insights into apoL1 and TLF mode of action (see figure below). This approach is not possible in Leishmania and T. cruzi, due to the absence of the RNAi pathway. However, selection of the T. brucei RNAi library in the anti-T. cruzi and anti-leishmanial drugs has enabled us to identify panels of candidate efficacy determinants with orthologues in T. cruzi and Leishmania (Collett et al, Antimicrobial Agents and Chemotherapy, 2019).
Selective screens, such as those described above, are only the first stage of the process, and not an end in themselves. The roles and contributions of the key proteins identified in each screen are validated using a variety of techniques available in the lab, depending on the character of the protein(s) under investigation. These include phenotypic analysis following specific stem-loop RNAi or gene deletion, tagged-protein localisation, and over-expression of wild type and mutant proteins, all of which provide insights into protein function. All these approaches are enabled by the suite of molecular tools that we have in the lab (these are freely available to the wider T. brucei research community on request)